Research Articles
Validating Computational Chemistry Predictions: A Practical Framework for Researchers and Drug Developers
This article provides a comprehensive framework for validating computational chemistry predictions, crucial for ensuring reliability in drug design and materials science.
Benchmarking in Computational Chemistry: A Guide to Validating Models for Drug Discovery and Biomedical Research
This article provides a comprehensive guide to benchmarking in computational chemistry, a critical process for validating the accuracy and reliability of models that predict molecular properties and behaviors.
Validating Multi-Level Quantum Chemistry Workflows: A 2025 Roadmap from Theory to Clinical Application
This article provides a comprehensive framework for the validation and comparison of multi-level quantum chemistry workflows, a critical frontier in computational drug discovery.
Benchmarking Quantum Chemistry: How the IHD302 Set Challenges Dimerization Energy Convergence
This article explores the performance and implications of the IHD302 benchmark set, a comprehensive collection of 604 dimerization energies for 302 inorganic heterocycles composed of p-block elements.
Convergence Challenges and Solutions: Assessing Quantum Chemistry Methods for Heavy Elements
Accurately modeling the electronic structure of heavy elements remains a formidable challenge in quantum chemistry due to strong electron correlation and significant relativistic effects.
Convergence Methods for Open-Shell Systems: A Comparative Analysis for Computational Chemistry and Drug Discovery
This article provides a comprehensive comparative analysis of convergence methods for high-spin open-shell systems, a critical challenge in computational chemistry and computer-aided drug discovery (CADD).
Validating SCF Convergence Protocols for p-Block Elements: A Quantum Chemistry Benchmarking Guide
Accurate self-consistent field (SCF) convergence for p-block elements is critical for reliable quantum chemical predictions in drug design and materials science.
Benchmarking GFN Methods: Accuracy and Efficiency for Organic Semiconductor Design
This article provides a comprehensive assessment of the semiempirical GFN method family (GFN1-xTB, GFN2-xTB, GFN0-xTB, and GFN-FF) for modeling organic semiconductors.
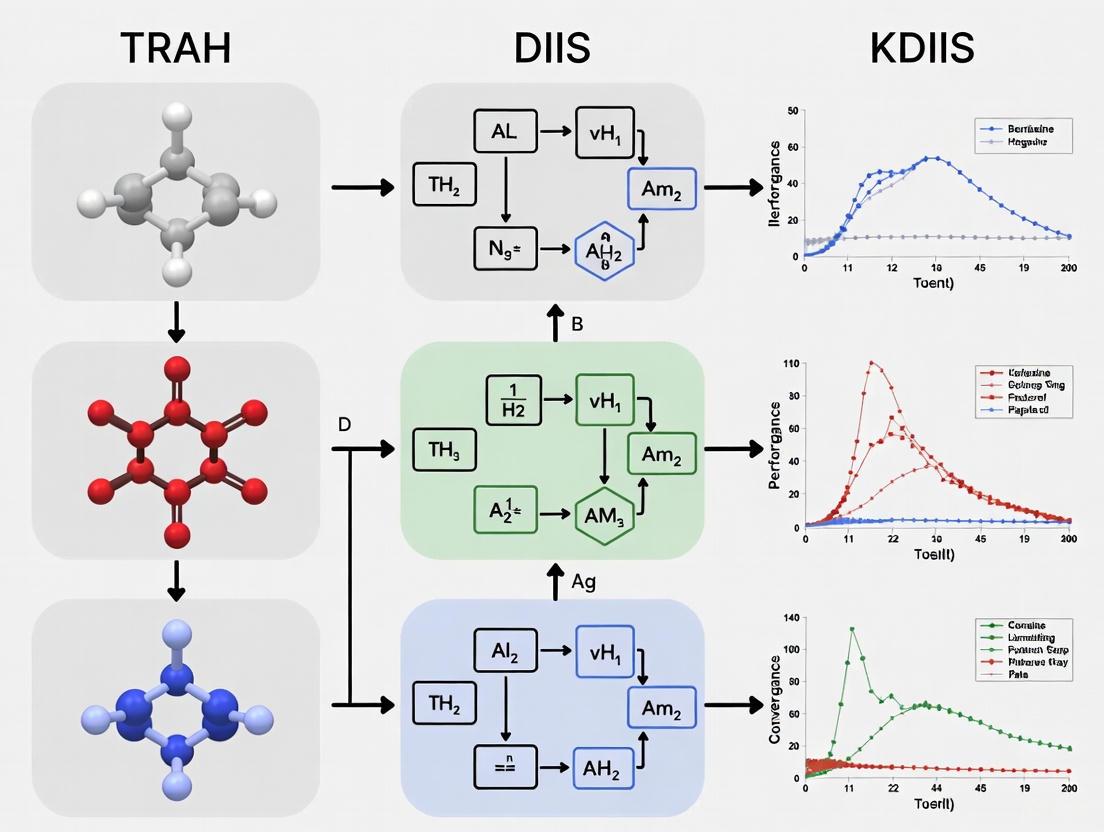
TRAH vs DIIS vs KDIIS: A 2025 Guide to SCF Convergence Performance for Computational Drug Discovery
This article provides a comprehensive comparison of three central Self-Consistent Field (SCF) convergence algorithms—TRAH, DIIS, and KDIIS—tailored for researchers and developers in computational chemistry and drug discovery.
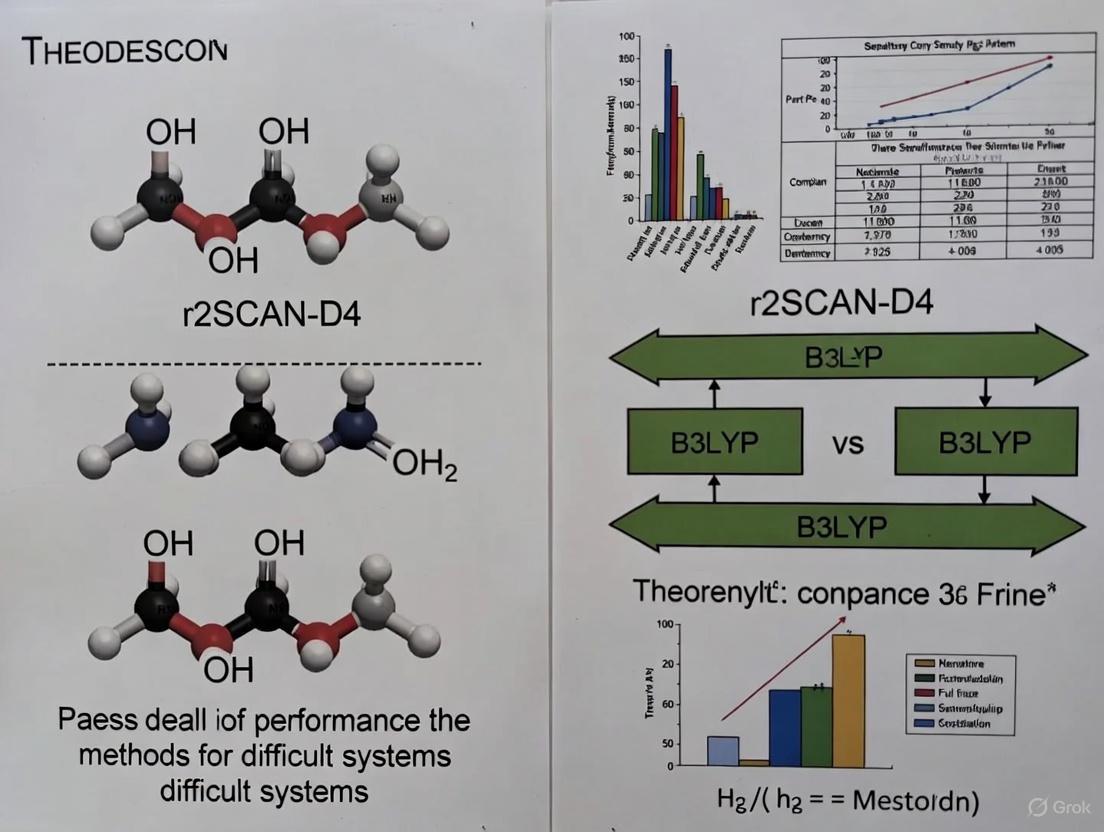
r2SCAN-D4 vs. B3LYP: A Performance Benchmark for Challenging Chemical Systems in Drug Development
This article provides a comprehensive benchmark analysis of the modern meta-GGA functional r2SCAN-D4 against the widely used B3LYP, focusing on their performance for difficult systems relevant to pharmaceutical research.